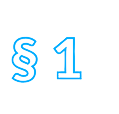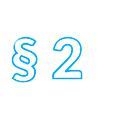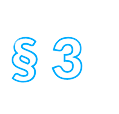Die Richtlinie 2001/83/EG‚ ist eine EG-Richtlinie Richtlinie der Europäische Union Europäischen Union, in der die seit 1965 verabschiedeten Richtlinien, die Humanarzneimittel betreffen, zu einem Gemeinschaftskodex zusammengefasst wurden. Durch eine Reihe von EG-Richtlinien ist in den letzten Jahrzehnten das Arzneimittelrecht in der Europäischen Union weitgehend harmonisiert worden. Als Richtlinie des Europäisches Parlament Europäischen Parlaments und des Europäischer Rat Rates wirkt die Richtlinie 2001/83/EG mittelbar, indem sie die Mitgliedstaaten der Europäischen Union verpflichtet, die Richtlinie in nationale Gesetze umzusetzen.
Bedeutung
Die Richtlinie 2001/83/EG kodifiziert Grundsätze zur Herstellung, Zulassung, zum Inverkehrbringen und zur Überwachung von Humanarzneimitteln. Damit sind zur Verwirklichung des Europäischen Binnenmarktes für Arzneimittel technisch-wissenschaftliche Hürden abgebaut worden. Durch die Richtlinie werden unter anderem die nicht zentralisierten Verfahren zur EU-weiten Arzneimittelzulassung definiert. Bei Bedenken einzelner Mitgliedstaaten wegen einer schwerwiegenden Gefahr für die öffentliche Gesundheit sind Schiedsverfahren vorgesehen. Von der Harmonisierung sind sozialrechtliche Aspekte wie die Preisfestsetzung und Erstattung von Arzneimitteln nicht berührt.
Analog zur Richtlinie 2001/83/EG wurden alle Richtlinien, die Arzneimittel zur Anwendung bei Tieren betrafen, in der Richtlinie 2001/82/EG zu einem Gemeinschaftskodex für Tierarzneimittel zusammengefasst.
Das zentralisierte EU-Zulassungsverfahren wird nicht durch die Richtlinie sondern durch die Verordnung EG Nr. 726/2004 bestimmt.
Umsetzung in nationales Recht
In Österreich ist die Richtlinie mit dem Arzneimittelgesetzund der Arzneimittelbetriebsordnung umgesetzt worden.
Gliederung
Die Richtlinie ist in fünfzehn Titel und drei Anhänge gegliedert. Titel I enthält Begriffsbestimmungen, Titel II definiert das Anwendungsgebiet. Titel III regelt das Inverkehrbringen, Titel IV die Herstellung und den Import von Arzneimitteln. Titel V befasst sich mit der Etikettierung und der Packungsbeilage, Titel VI mit der Einstufung von Arzneimitteln, unter anderem in der Verschreibungspflicht. Titel VII regelt den Großhandel, Titel VIIa den Verkauf im Fernabsatz, Titel VIII die Arzneimittelwerbung, der Titel VIIIa nochmals Arzneimittelinformation und Werbung. Titel IX enthält Regelungen zur Pharmakovigilanz, Titel X zu Blutprodukten. Titel XI ist mit Überwachungen und Sanktionen befasst. Titel XII ist die Rechtsgrundlage für den Ständigen Ausschuss, in dem Vertreter der Mitgliedstaaten die Europäische Kommission bei der Anpassung der Richtlinien, aber auch bei Entscheidungen zu Arzneimittelzulassungen unterstützen. Schließlich enthält Titel XIII allgemeine Bestimmungen und Titel XIV Schlussbestimmungen.
Im umfangreichen Anhang I werden detailliert die Zulassungsanforderungen für Humanarzneimittel aufgeführt. Teil I enthält die Standardanforderungen, darunter die Nachweise zur pharmazeutischen Qualität, zur nichtklinischen Pharmakologie und Toxizitätsbestimmung sowie zur klinischen Prüfung. Ferner enthält der Teil I Angaben zum Common Technical Document, dem Format, in dem die Zulassungsunterlagen einzureichen sind. Teil II enthält Angaben zu spezifischen Zulassungsanträgen beispielsweise für Generika und Biosimilars. Teil III schreibt vor, welche Angaben zu speziellen Arzneimitteln, darunter biologischen beispielsweise Impfstoffe, radiopharmazeutischen, homöopathischen und phytotherapeutischen Arzneimitteln vorzulegen sind. Teil IV schließlich beschreibt die erforderlichen Angaben zu Arzneimitteln für neuartige Therapien, worunter Arzneimittel zur Gentherapie und zur somatischen Zelltherapie fallen.
Anhang II listet die durch die Richtlinie 2001/83/EG aufgehobenen Richtlinien auf, nebst Fristen, mit denen diese in den Mitgliedstaaten in nationales Recht umzusetzen waren. Anhang III enthält eine Entsprechungstabelle zwischen der neuen Richtlinie und den aufgehobenen Richtlinien.
Weblinks
- http://www.pei.de/SharedDocs/Downloads/gesetze/richtlinie-2001-83-eg-gemeinschaftskodex-humanarzneimittel-2001-11-06.html?nn=3487394 Paul-Ehrlich-Institut: Konsolidierte Fassung der Richtlinie auf dem Stand bis zu der Berücksichtigung des Artikels 1 der Richtlinie 2011/62/EU des Europäischen Parlaments und des Rates vom 8. Juni 2011 PDF; 1093 kB
Quellen
http://de.wikipedia.org/wiki/Richtlinie_2001/83/EG Gemeinschaftskodex f%C3%BCr Humanarzneimittel 18.12.2014
Lizenzinformation zu diesem Artikel
Dieser Artikel basiert auf dem in den Quellen angeführten Wikipedia-Artikel, verfügbar unter der Lizenz „CC BY-SA 3.0„.